Síndrome de Angelman
El síndrome de Angelman es un trastorno neurológico crónico de origen genético o epigenético en el ser humano. Existen un gran número de alteraciones genéticas que pueden afectar al desarrollo del feto humano. La mayoría de ellas impiden que el embarazo llegue a término, aunque algunas de ellas permiten la supervivencia del individuo. El síndrome de Angelman permite que nazca el bebé, aunque éste presentará graves problemas de desarrollo tanto cognitivos como de coordinación motriz. Se estima que uno de cada veinte mil embarazos puede sufrir los efectos de este trastorno genético.
Causas: los estudios genéticos han descubierto que esta dolencia neuronal se debe a errores en el gen UBE3A, que codifica para una ubiquitin ligasa denominada E3A. Este gen se encuentra en el cromosoma 15, en la misma región que es la responsable del síndrome de Prader Willi, del que puedes saber más en su propio artículo aquí.
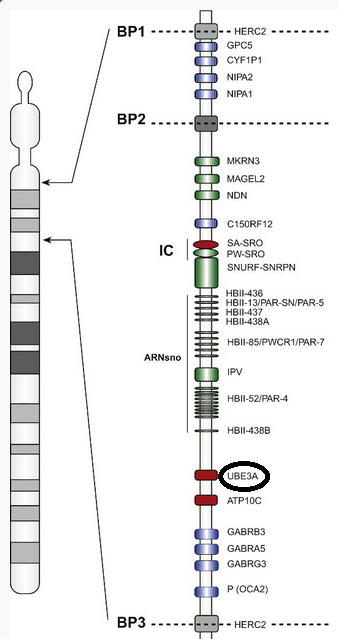
El gen UBE3A se encuentra en el cromosoma 15, cambios en su expresión provocan el síndrome de Angelman.
Este síndrome está causado por fallos en la impronta genética del cromosoma 15 materno, o la deleción de la región 15q11-q13 (la región entre los puntos 11 y 13 del brazo corto del cromosoma 15 materno). Puedes leer más sobre la impronta genética aquí. La enfermedad se desarrolla cuando el gen del cromosoma de origen materno del feto no se expresa. Mientras que si no se expresa el de origen paterno se produce el síndrome de Prader Willi.
Síntomas: los síntomas del síndrome de Angelman son muy variados y su intensidad o presencia varía entre los individuos y durante la edad del afectado. Los síntomas más comunes son una discapacidad intelectual entre severa y moderad, algunos pueden llegar a hablar. Aunque rara vez tienen unas habilidad social básica. El desarrollo está muy retrasado, durante la edad adulta todavía conservan algunos rasgos infantiles. Casi nunca pueden mantenerse erguidos solos y acaban presentando ataxia muchas veces, por lo que los adultos rara vez son capaces de andar. Una de las características más destacadas es la apariencia de felicidad constante, siempre sonrientes, siendo muy fácilmente excitables. Presentan hiperactividad e insomnio, que se acentúa todavía más en la edad adulta, llegando a dormir menos de 5 horas diarias.
Además encontramos otros síntomas menos comunes como la espina bífida, dificultades gástricas severas, problemas para digerir o masticar, frecuentes babeos. Estos individuos parecen además propensos a la obesidad, aunque bien es verdad que no son capaces de realizar muchos ejercicios, por su falta de coordinación motriz.
Diagnóstico, tratamiento y pronóstico: El diagnóstico se lleva a cabo mediante el cariotipo del feto, donde se puede observar si hay deleciones en los cromosomas. Aunque a veces son necesarios estudios epigenéticos para detectar esta dolencia. La enfermedad no tiene cura, aunque algunos de sus efectos pueden ser controlados de forma específica, como por ejemplo la epilepsia o la escoliosis. El pronóstico de los individuos depende del grado de afectación de la enfermedad, la localización de la mutación, la efectividad de la impronta, o el tamaño de la deleción son los responsables de las diferencias en la expresión de la proteína correcta. Algunos de ellos pueden presentar un alto grado de conciencia y los adultos pueden llegar a desarrollar una funcionalidad reducida.